Protocols
7-AAD ON FIXED CELLS
Stain dead cells and then fix them so that only the dead cells keep the 7-AAD
styles - DO NOT REMOVE
More Info on 7-AAD
FLUORESCENT DETECTION OF NON-VIABLE CELLS IN FIXED CELL PREPARATIONS
T. J. Fetterhoff, S.P. Holland, and K. J. Wile, Boehringer Mannheim Corp., Research & Development, Indianapolis, IN, USA. Cytometry Supplement 6: 27 (1993).
Cell lysis and or fixation have become common practice in flow cytometric sample preparation. A procedure has been developed that allows detection of cells that were non-viable prior to cell fixation. This technique overcomes the problems of artifacts caused from non-specific uptake of antibodies by non-viable cells at the time of staining. 7-Amino-Actinomycin D (7-AAD) is used to identify non- viable cells during immunostaining. Following several washes, cells are lysed or fixed in solutions that contain molar excess of the non-fluorescent Actinomycin D (AD). AD competes with 7-AAD for binding sites to dsDNA and inhibits further uptake by 7-AAD. Furthermore, 7AAD exhibits spectral characteristics enabling fluorescence separation from FITC as well as PE. This procedure allows cell fixation to inactivate infectious agents and eliminates artefactual staining by non-viable cells. © 1993 Wiley-Liss, Inc.
Materials
- 7-Amino-Actinomycin D (Calbiochem Cat #129935 )
- Actinomycin D (Alexis Biochemicals Cat # ALX-380-009 )
- Phospahte Buffered Saline (PBS)
- Para-formaldehyde, (From 2% Stock)
Protocol
- Resuspend labeled cells in 100 ml PBS that contains 20 mg 7-AAD; incubate for 30 minutes at 4°C.
- Centrifuge and resuspend pellet in 100 ml of PBS that contains 0.5% para-formaldehyde and 50 mg actinomycin-D; incubate 30 minutes at 4°C
- Centrifuge and resuspend pellet in PBS; hold at 4°C in the dark.
References
- Schmid I, Uittenbogaart CH, Krall WJ, Braun J and Giorgi JV. Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 13:204-208, 1992.
- Fetterhoff TJ, Holland SP, and Wile, KJ. Fluorescent detection of non-viable cells in fixed cell preparations. Cytometry 14 (Suppl. 6):27, 1993.
ADHERENT CELL DNA STAINING
Only use if the other protocols fail. The samples are stored before staining, but it's more complicated.
styles - DO NOT REMOVE
DNA Content of Adherent Cell
Reagents
CRYOPRESERVATIVE:
- 250 mM sucrose
- 5% dimethylsulfoxide (DMSO) v/v
- 40 mM trisodium citrate
- pH 7.6
STOCK SOLUTION:
- 3.5 mM trisodium citrate
- 0.1% v/v Nonidet P-40
- 0.5 mM TRIS
- 1.5 mM spermine tetrahydrochloride
- pH 7.6
RNASE SOLUTION:
- 500 µg/ml trypsin inhibitor
- 10 µg/ml RNAse
PROPIDIUM IODIDE (PI) SOLUTION:
- 42 µg/ml PI
- 1.16 mg/ml spermine tetrahydrochloride
Procedure
- Remove media from culture dish and rinse cells with 1.0 ml of PBS.
- Add 500 µl of Cryopreservative.
- Store dishes at -80°C until ready to stain.
- Thaw at room temperature and remove Cryopreservative.
- Add 900 µl Trypsin Solution (30µg/ml trypsin in Stock Solution).
- Mix gently and keep at room temperature for 10 min.
- Add 750 µl RNAse Solution.
- Mix gently and keep at room temperature for 10 min.
- Add 750 µl PI Solution.
- Filter solution through Falcon® Cell Strainer Cap (Cat. No. [35]2235) and keep on ice protected from light for at least 30 min & up to 3 hours.
Reference
Tennenbaum T, Giloh H, Fusenig NE, Kapitulnik J: A rapid procedure for the flow cytometric DNA analysis in cultures of normal and transformed epidermal cells. Journal of Investigtive Dermatology, 90:857-860, 1988.
ANTIBODY STAINING
General Antibody Staining Protocol
styles - DO NOT REMOVE
Basic Antibody Staining
This protocol is for cultured cells.
For each monoclonal antibody (MAb) marker use 1 X 105 - 1 X 107 cells per 100 µl volume:
- If adherent cells, trypsinize, scrape or treat with EDTA to get single cells suspension, otherwise skip to step 2.
- Wash cells with 2 ml COLD Phosphate Buffered Saline (PBS) or another buffer with 0.1%-5% protein (eg Bovine Serum Albumin, BSA) by centrifuging at 150-300 X g in the COLD (4°C).
- Wash once more (Optional).
Staining
- Aliquot 100 µl of 1-100 X 105 cells (use wash buffer above with 0.1% NaN3 added) in Falcon® #2052 or #2054 tubes on ice.
- Add 20 µl MAb (1-10 µg/ml final concentration, depending on Mab).
- Incubate 10 - 30 min on ice.
- Wash again as above with 2 ml cold azide buffer.
- Resuspend in 100 µl of Secondary Antibody (eg. Fluorescinated Goat anti-mouse IgG). Otherwise skip to step 7 if using directly conjugated MAb.
- Incubate 10 - 30 min on ice in DARK. Wash again with azide buffer.
- Resuspend in 100 to 500 µl COLD wash buffer and add equal volume of COLD buffered 2% para-formaldehyde (see recipe) to fix cells. OR leave in wash buffer and analyze LIVE (Add propidium iodide, 0.5 µg/ml final concentration). Ideal final concentration of cells should be 1 X 106/ml.
APOPTOSIS WITH PI ONLY
To stain cells with only PI to measure apoptosis (quick and dirty)
Detection of Apoptosis by Propidium Iodide Only
Reagents
FIXATIVE:
- Hank's Balanced Saline Solution (HBSS, Sigma H1387 or H8264)
- 70% EtOH (-20°C)
EXTRACTION BUFFER:
- 45 mM Na2HPO4
- 2.5 mM citric acid
- 0.1% Triton X-100
- pH 7.8
STAINING SOLUTION:
- 10 mM PIPES (Sigma P1851)
- 100 mM NaCl
- 2 mM Mg2Cl
- 0.2 % Triton X-100
- 50 µg/ml Propidium Iodide (PI)
- 50 units/ml RNAse (1:17.2 of 10 mg/ml, DNAse-free)
- pH 6.8
Fixation
Add dropwise 2 X 106 cells in 500 µl of ice-cold HBSS to 4.50 ml of 70% EtOH at -20°C while slowly vortexing. Do not substitute PBS for HBSS.
Cells should be stored for at least 24 hrs at 4°C. Cells can be stored at -20°C for at least 2 weeks.
Extraction
- Centrifuge tubes at 400 X g for 10 minutes, then aspirate supernatant.
- Resuspend pellet in 200 µl of Extraction Buffer.
- Incubate for 20 min at 37°C.
Volume or time may need to be varied for optimal extraction.
Optional: Wash cells by centrifugation (as above in HBSS) before and/or after Extraction. If there is extensive fragmentation, Extraction Buffer may need to be diluted with HBSS to prevent over extraction.Stain
- Add 300 µl Staining Solution to above cell suspension.
- Transfer to Falcon® 12 X 75 mm Polystyrene round-bottom tubes (Cat #[35]-2054) if different tubes were used above. Falcon® Cat. No. [35]2235 have nylon filter caps and will remove clumps.
- Incubate 30 minutes room temperature, then place on ice bath protected from light.
Notes
- Sub G1 peak can also represent mechanically damaged cells.
- Post-G1 cells that lose DNA may still have greater DNA content than G1 cells.
- Detergent permeabilization can be used instead of fixation.
- Formaldehyde fixation (cross-linking) precludes low molecular weight DNA extraction.
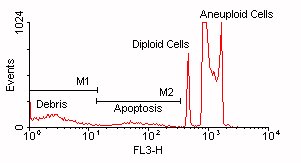
Click above figure for larger version (35,166 bytes)
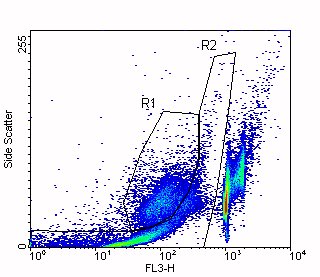
In this figure, you can see that the debris and apoptotic cells overlap in the DNA Content parameter (FL3-H). Region R1 indicates the apoptotic cells and Region R2 indicates the debris.
References
- Darzynkiewicz,Z, Flow Cytometry: Methods in Cell Biology, Volume 41, page 27 1994.
- Hotz, MA, Gong, J, Traganos, F, Darzynkiewicz, Z, Flow Cytometric Detection of Apoptosis: Comparison of the Assays of In Situ DNA Degradation and Chromatin Changes, Cytometry 15:237-244 (1994).
CELL CYCLE WITH PI
First choice protocol to stain most cell types with Propidium Iodide (PI) to measure DNA content
Cell Cycle Analysis by DNA Content (Propidium Iodide)
Fixation
- Wash cells by centrifugation (e.g. 200 x g, 5 min, 4°C) in protein-free buffer, such as Phosphate Buffered Saline without Ca+2 or Mg+2 (PBS).
- (Optional) Repeat step 1.
- Resuspend at 2 x 106 cells in 1 ml ICE COLD BUFFER. Cell number will effect staining quality!Optional: Use pre-coated or silanized polypropylene tubes to minimize sticking. Pre-coat tubes overnight with 2% Bovine Serum Albumin (BSA) in PBS.
- Vortex gently, slowly adding the cell suspension dropwise to 9 ml of 70% ethanol in a 15 ml polypropylene centrifuge tube (Falcon® Cat. No. [35]2097). OR: Vortex gently, slowly adding the cell suspension dropwise to an equal volume of COLD ABSOLUTE ethanol. Optional: Observe cell preparation with a microcope to verify minimum cell clumping.
- Store at 4°C to - 40°C for AT LEAST 2 hours, 12 - 24 hours is best. Can be stored for up to 2 years before staining.
- Centrifuge cells at 200 x g, 10 min, 4°C.
- Resuspend pellet in 3 ml COLD PBS and transfer to Falcon® 12 X 75 mm (Cat. No. [35]2054) polystyrene tubes for staining if other tubes (polypropylene) were used for the fixation steps above. Falcon® Cat. No. [35]2235 have nylon filter caps and will remove clumps.
Staining with Propidium Iodide (PI)
- Wash cells at least once with COLD PBS. Cells may form a diffuse ring-shaped pellet, so centrifuge longer ( e.g. 200 x g, 10 min, 4°C).
- Resuspend cells in 300 - 500 µl PI/Triton X-100 staining solution: to 10 ml of 0. 1 % (v/v) Triton X-100 (Sigma) in PBS add 2 mg DNAse-free RNAse A (Sigma) and 0.40 ml of 500 µg/ml PI (e.g., Roche). Prepare freshly. A stock solution of PI, made by dissolving 1 mg PI in 2 ml water, can be stored several months at 0° to 4°C. (Or buy 500 µg/ml PI from Roche new Catalog # 11348639001, old Cat. No. 1348639). Note: If the RNAse is not DNAse-free, boil a solution of 2 mg RNAse A in 1 ml water for 5 min. Aliquot and store at -20°C.
- Incubate 37°C for 15 minutes or for 30 min at 20°C.
- Transfer tubes to ice or store at 4°C PROTECTED FROM LIGHT.
- Acquire data on flow cytometer within 48 hours (but might last up to 2 weeks). May require nylon mesh filtration (eg, Filcons, BD Cat. No. 340627) to remove cell clumps or syringing (25 gauge, UCSD Storehouse # 7245) to break up cell clumps. Can acquire 5-30 samples per hour, depending on cell preparation.
- MulticycleAV (IBM-PC) or ModFit LT (Macintosh) is used to fit the data to various cell cycle models. See below for examples
THIS A SCREEN SHOT OF A TYPICAL PROFILE FROM MULTICYCLEAV:
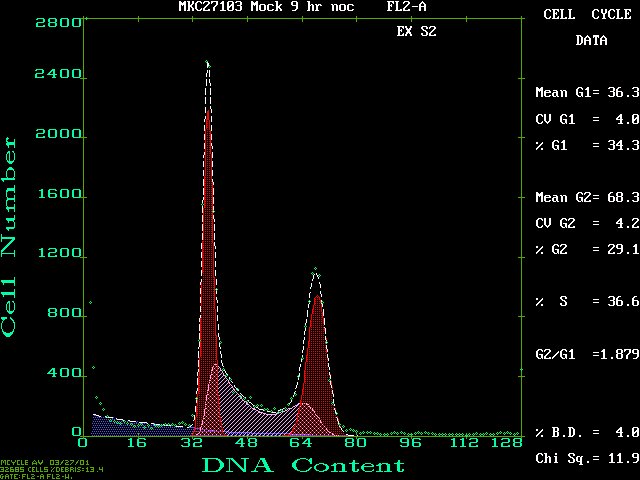
Click on this figure to see the full screen.
HERE'S A MODFIT LT OUTPUT EXAMPLE FROM THE SAME FCS FILE:
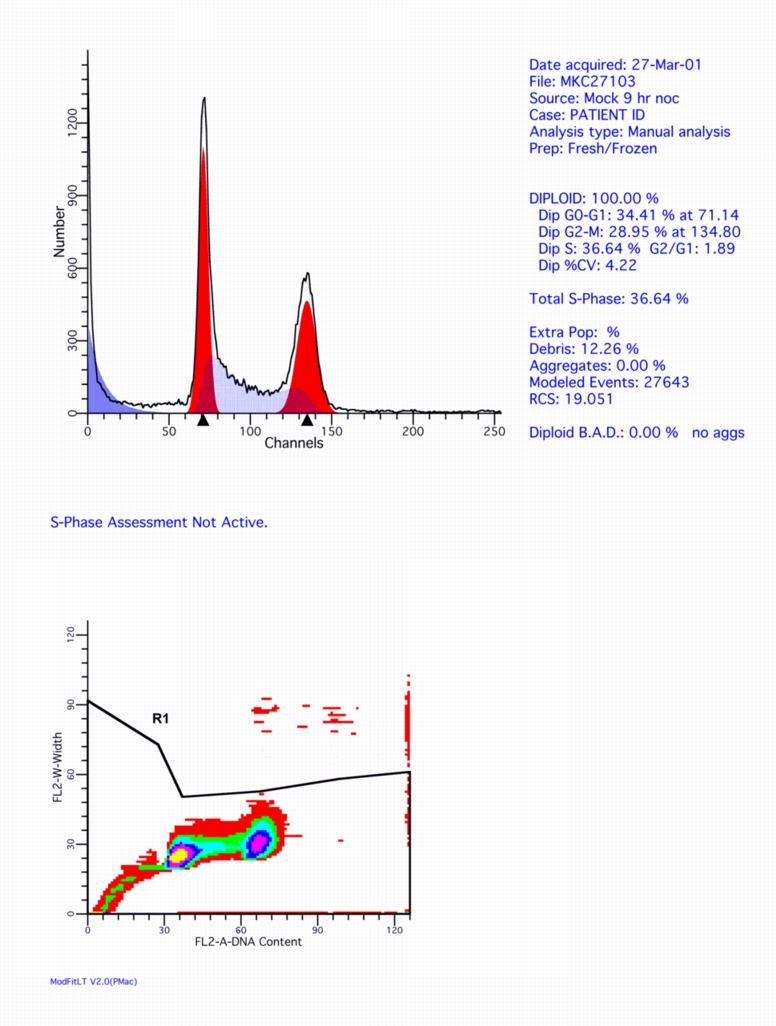
Click on this figure to see the full screen.
References
- Shapiro, HM, Practical Flow Cytometry, second edition. New York: Alan R. Liss, Inc; 1988. 353 p.
- Darzynkiewicz, Z, Nucleic Acid Analysis. In: Robinson, JP, managing editor. Current Protocols in Cytometry. New York : J Wiley & Sons, Inc; 1997. Chapter 7.
DNASE I
DNAse treatment of cells that tend to clump
More Info on DNASE
Cell clumping can result in poor sort purity when sorted target cells are attached to non-target cells, and poor recovery when coincident aborts exclude all clumped cells. DNA from lysed cells in the medium can cause cells to clump.
Materials
- DNAse (Sigma D-4513) 100 µg/ml in Hank's Balanced Salt solution (HBSS, Sigma H-6648)
- Magnesium chloride hexahydrate (Sigma M-2670) MW=203.3
- 203 mg/ml = 1000 mM or 200X
Procedure
- Treat cells for 15 to 30 minutes in a solution of 100 µg/mL DNAse and 5 mM MgCl2 in HBSS at room temperature.
- Wash the cells once in the presence of 5 mM MgCl2 in HBSS.
- Filter through Falcon® Cell Strainer (Catalog #[35]2340 or #[35]2235).
- Gently suspend the cells Stain Buffer (BSA) containing MgCl2 and 25-50 µg/mL DNAse (as a maintenance dose) prior to and during the sort.
Notes
DNAse I requires a concentration of at least 1 mM magnesium to work effectively, although 5 mM is optimal.
It is important to minimize the presence of dead cells during this procedure, since actin released from dead cells irreversibly inhibits DNAse I.
References
Crissman, HA, Mullaney, PF, and Steinkamp, JA. Methods and applications of flow systems for analysis and sorting of mammalian cells. Meth. Cell Biol. 9:175 (1975).
ETHIDIUM MONOAZIDE (EMA)
Stain dead cells and then be able to fix them, as with the 7-AAD above.
More Info on EMA
Adapted from Kathrine A. Muirhead
Zynaxis Cell Science, Inc.
- Use ethidium monoazide (EMA) that has been titered (0.5 ug/ml - 10 ug/ml) and compared to trypan blue exclusion and/or propidium iodide (PI = 0.5 ug/ml). Viable cells should have low background EMA staining.
- Wash cells twice in phosphate buffered saline (PBS) containing 0.5% to 5% bovine serum albumin (BSA) and 0.1% sodium azide (NaN3).
- Resuspend cells to 107 per ml.
- Add 50 ul cells and appropriate volume of buffer to give 90 ul in 96-well plate or 12 x 75 mm tubes. Add sufficient EMA stock to each well or tube for the final concentration (as determined above) and mix well. (eg 10 ul of 100 ug/ml EMA in PBS).
- Incubate IN THE DARK on ice for 15 minutes to allow uptake of EMA by dead cells.
- Continue incubating ON ICE for 15 minutes 30 cm from fluorescenct light source to photoactivate EMA and cause covalent binding.
- Add appropriate amount of monoclonal antibodies and incubate 15 - 30 minutes on ice in the dark.
- Wash twice with wash buffer (eg PBS-BSA-NaN3).
- Fix cells with 0.5% final concentration protein-free buffered paraformaldehyde (PFA) and store at 4°C IN DARK.
GFP AND DNA
Determine the cell cycle of GFP positive and/or negative cells. Unfortunately, GFP usually has a 48 half-life!
Measurement of GFP and DNA Content in Fixed Cells
Materials
- Cells to be studied expressing green fluorescent protein (GFP). Note that the same cell type without GFP is needed as a control.
- Phospahte Buffered Saline (recipe link for PBS)
- Para-formaldehyde, (recipe link for 2% Stock)
- 70% Ethanol
- Propidium iodide stock solution (0.5 mg/ml in PBS)
- RNAse>
- 12 X 75 mm culture tubes
- Vortex mixer
- Ice bath
- Water bath at 37C
Protocol
FIX CELLS WITH FORMALDEHYDE
- Count cells.
- Place approximately 2 X 106 cells into a 12 x 15 mm test tube and wash them once with PBS by centrifugation for 5 min at 300 x g at 4°C.
- Remove supernatant by aspiration and add 500 µl of cold PBS to the cell pellet. Mix gently. Add 500 µl of ice cold, buffered 2% formaldehyde solution and mix again. Incubate at 4°C for 10 minutes to 1 hour.
Lower concentrations of formaldehyde and short incubation times preserve more fluorescence.
PERMEABILIZE CELLS WITH ETHANOL
- Spin cells down by centrifugation for 5 min at 300 x g at 4°C, remove supernatant by aspiration, wash once with cold 1 X PBS, then add 1 ml of 70% ethanol at -20°C drop-wise to the cell pellet with the tube sitting on a vortex. Incubate cell suspension overnight at 4°C.
STAIN WITH PROPIDIUM IODIDE
- Spin cells down by centrifugation for 5 min at 300 x g at 4°C, remove supernatant by aspiration and add 1 ml of a solution containing 40 µg/ml of PI and 100 µg/ml of RNAse. Incubate cell suspension at 37°C in the dark for 30 min.
- Transfer tubes to ice bath. If needed, filter samples through a nylon mesh (BD Cat #35-2340 or 35-2235) to remove clumps before acquisition on the flow cytometer.
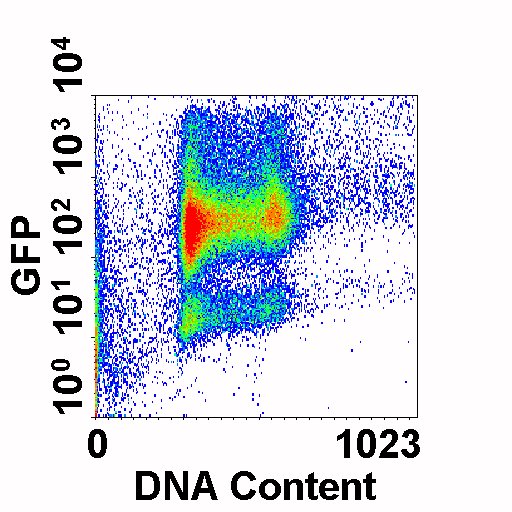
This is an example of cells expressing varying levels of GFP (y-axis) versus DNA Content (PI, x-axis)
References
- Chu, YW, Wang R., Schmid I, Sakamoto KM. Analysis with flow cytometry of green fluorescent protein expression in leukemic cells. Cytometry 36:333-339, 1999.
- Schmid I. and Sakamoto KM. Analysis of DNA content and green fluorescent protein expression. In: Current Protocols in Cytometry, Vol 1, Robinson JP, Darzynkiewicz Z, Dean P, Orfao A, Rabinovitch P, Stewart C, Tanke H, Wheeless L, eds., John Wiley & Sons, 2001, pp. 7.16.1-7.16.10.
ISOLATED NUCLEI DNA STAINING
A quick way to stain suspension or adherent cells without storage
More Info on DNA Staining
Nuclei for Cell Staining Analysis
Hypotonic Staining Buffer:
| Sodium citrate tribasic dihydrate | 10.0 mg |
| Triton® X-100 | 30.0 ul |
| Propidium iodide | 5.0 mg |
| Ribonuclease A | 0.2 mg |
| H20 | 100 ml |
- Aliquot 1x106 cells into each tube.
- Centrifuge 200 to 400 xg for 5 - 10 minutes and aspirate supernatant without disturbing the pellet.
- Add 500 ul of the Hypotonic Staining Buffer to the pellet and gently mix well.
- Keep samples at 4°C protected from light for 30 min or for a maximum of 1 h before acquisition on the flow cytometer.
NOTE:
Debris can increase with longer staining incubation times.
Reference
Krishan A: Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 66:188-193, 1975.
NUCLEAR ANTIGEN AND DNA CONTENT WITH PI (2)
Use one of these protocols to stain cells for a nuclear antigen and DNA content
More Info on Antigen and DNA Content
These washless procedures require proper titration of antibodies.
Procedure A ( frozen cells)
- If adherent, trypsinize and wash once with PBS (without Ca++ and M++, with 2% Bovine Serum Albumin)
- Pellet cells and resuspend in ice-cold Freezing Buffer to a minimum concentration of 105 cells in 50 ml and store at –80°C
- While thawed cells are slowly agitated in an ice bath mounted on a mixer, add 200 ml Lysis-DNA Staining Solution and mix for 15 minutes
- Add 25 ml FITC-conjugated antibody and mix for 30 minutes
- Analyze
REAGENTS:
- Freezing buffer:
250 mM sucrose
40 mM sodium citrate
5% v/v DMSO
pH 7.6 - Lysis-DNA staining solution:
0.5% Nonidet P40
20 mg/ml propidium iodide (PI)
0.2 mg/ml RNAse
0.5 nM EDTA
pH 7.2
Procedure B (fresh cells)
- Suspend 2 x 105 fresh cells in PBS and mix with 100 ml Lysing Solution supplemented with appropriate dilution of antibody and incubate 30 min at room temperature.
- Add 100 ml of an appropriate dilution of the secondary FITC-conjugated antibody containing PI (final concentration 10 mg/ml) and RNAse (final concentration 100 mg/ml) and incubate 30 minutes RT.
REAGENT:
- Lysing solution:
1% Bovine Serum Albumin (BSA)
0.5% Triton X-100
0.2 mg/ml EDTA
in PBS
References
- Larsen, J.K., “Washless” Procedures for Nuclear Antigen Detection. In: Methods in Cell Biology: Flow Cytometry, Vol. 41, Chapter 24, page 377-388. Academic Press, Inc. San Diego. 1994
- Landberg, G., Roos, G. Flow cytometric analysis of proliferation associated nuclear antigens using washless staining of unfixed cells. Cytometry, 1992, 13(3):230-40.
- Vindelov, L.L., Christensen, I.J., Keiding, N., Spang-Thomsen, M., Nissen, N.I., Long-term storage of samples for flow cytometric DNA analysis. Cytometry 3 (1983), 317-322.
- Baisch, H., Gerdes, J. Simultaneous staining of exponentially growing versus plateau phase cells with the proliferation-associated antibody Ki-67 and propidium iodide: analysis by flow cytometry. Cell and Tissue Kinetics, 1987 Jul, 20(4):387-91.
- Palutke M; KuKuruga D; Tabaczka P. A flow cytometric method for measuring lymphocyte proliferation directly from tissue culture plates using Ki-67 and propidium iodide. Journal of Immunological Methods, 1987 Dec 4, 105(1):97-105.
- Sasaki K; Murakami T; Kawasaki M; Takahashi M. The cell cycle associated change of the Ki-67 reactive nuclear antigen expression. Journal of Cellular Physiology, 1987 Dec, 133(3):579-84.
- Drach J; Gattringer C; Glassl H; Drach D; Huber H. The biological and clinical significance of the KI-67 growth fraction in multiple myeloma. Hematological Oncology, 1992 Mar-Apr, 10(2):125-34.
- Jacob MC; Favre M; Bensa JC. Membrane cell permeabilization with saponin and multiparametric analysis by flow cytometry. Cytometry, 1991, 12(6):550-8.
SURFACE ANTIGEN AND DNA CONTENT WITH PI
To stain surface antigens and measure DNA Content
Surface Antigens and DNA: Hypotonic Propidium Iodide Staining
This protocol is ideal for surface labeled cells.
Stock solutions (stored at 4°C)
1 mg/ml trisodium citrate
1 mg/ml RNAse in Phosphate Buffered Saline (PBS)
Note: You cannot boil PBS!
10% Triton X-100 (v/v in H2O)
500 mg/ml propidium iodide (PI Solution, Roche Cat. No. 1 348 639)
Working solution
(for 100 samples; prepare weekly, store at 4°C)
5.0 ml RNAse
500 ml 10% Triton X-100
2.0 ml PI Solution
42.5 ml trisodium citrate
Staining procedure
(after last antibody incubation)
- Wash 106 cells with protein-free PBS, aspirate supernatant and break up pellet.
- Incubate 10 min on ice.
- Add 500 ml ice cold Working Solution and mix gently.
- Incubate overnight on ice (in refrigerator).
Reference
Brons PPT, Van Erp PEJ, Pennings AHM. Flow Cytometry: Methods in Cell Biology, Volume 41, Chapter 6, page 97. (1994)
SORTING PREPARATION
This will help you prepare for sorting and estimate yields
Sorting Checklist
Sorting is by appointment only. Telephone (858) 822-0407 to set an appointment or for more information. You may need to use voicemail, but do not e-mail.
Sample
Provide tubes pre-filled with 4 ml of the sample buffer (no cells) for rinsing.
Suspend cells in PBS or HBSS with 5% (2 - 10%) BSA at up to 3 X 107 per ml in Falcon ® [35]2063 12 X 75 mm or [35]2097 17 X 120 polypropylene tubes.
- [35]2054 or [35]2058 12 X 75 mm polystyrene tubes can also be used.
- Serum as the carrier protein is NOT recommended, as it adds to the background fluorescence signal.
Particle concentration effects sort speed. About 1 ml/hr of sample can be processed:
- 7 X 106 TOTAL particles per ml is about 25 million per hour
- Use the highest concentration possible while maintaining a single cell suspension
- 20 - 100 million cells/ml
- We can dilute sample with your provided buffer at the sorter, but can't concentrate them.
- Use the highest concentration possible while maintaining a single cell suspension
Filter cells through
- Falcon® Cell Strainers ([35]2340) for large numbers of cells
- Cell-Strainer Caps ([35]2235) can also be useful
Some cell types may need to be treated with DNAse to remove dead cell clumps and may also need to be suspended in DNAse buffer to maintain a single cell suspension. See DNAse Protocol or use Accumax from Innovative Cell Technologies.
- An alternate solution to prevent aggregates is 1 mM EDTA.
- NOT compatible with DNAse!
Bulk Collection
Sorted cells should NOT be collected into empty tubes.
- 100% serum is recommended for bulk sorting.
- Tubes should be kept cold & in the dark.
FOUR populations can simultaneously be sorted into:
- 12 x 75 mm tube (Falcon® [35]2063 is the best)
- Use 1 ml serum per 12 X 75 mm tube
-
- Holds about 0.5 million sorted cells
- 1.5 ml Eppendorf tubes
- Cut the hinge!
- 1.2 ml microtiter tubes (Fisher #02-681-383)
TWO populations can be collected into tubes above or larger tubes
- eg, 17 X 120 mm tube Falcon® [35]2097)
- Use 1 ml serum per 17 X 120 mm tube
- Will hold about 2 million sorted cells
Automated Cell Deposition Unit (ACDU)
Single cells can be sorted at 1 - 50,000 particles:
- Onto a slide or into microtiter wells (6,12,24,48,96 or 384 wells per plate)
- Plates should be pre-filled (100 microliters conditioned media in 96 well plates) and pre-gassed.
- Keep in a sealed sandwich bag or keep a tiny chip of CO2 in the plate's container (large box) to keep bicarbonate-buffered media's pH correct (pink media is not good).
- Or use 50% serum in PBS and change buffer when you return to your lab.
- Can also directly sort into PCR extraction buffer or other non-culture buffer (i.e. serum)
- Plates should be pre-filled (100 microliters conditioned media in 96 well plates) and pre-gassed.
Cell cloning works best in phenol red-free media and/or on feeder cells
- 1,000 cells = 3.5 microliters sheath (PBS), of which only approximately 1% is the original sample solution
Sort Estimator
Example to estimate THEORETICAL yield (Y):
Yield = R*P/100*EXP(-R*n*T)
where T = 3.7*[SQRT(p)/4.5*(d)/106] = 69,490 Hz
AND
R = 6,800 cells per second
P = 1 percent positive
d = 70 micron nozzle
p = 35 psi
n = 1.5 drop packet
OR
58 cells per second (211,380 per hour), 86% recovery.
This does NOT count sort aborts.
TITRATION
To get the most out of your antibodies, titrate them.
Antibody Titration
Directly Labeled
- Determine concentration of Antibody (Ab) and centrifuge 10 min at 15,000 X g, 4° C to remove aggregates. Discard pellet.
- Start with Ab stock at 300 µg/ml in Phosphate Buffered Saline (PBS). Serial dilute six tubes:
10 µl Ab + 20 µl PBS (or 20 µl of previous dilution) = 30 µl
- Resuspend cells in 200 µg/ml normal IgG of detecting species at 5 - 10 X 106 cells/ml.
- Aliquot 50 µl of cells in (Falcon #2054) 12 X 75 mm tubes on ice.
- Add 10 µl of each Ab dilution. Also prepare a tube with cells alone (autofluorescence control) and one with the detecting species IgG negative control.
- Gently mix and incubate 15 - 45 min on ice IN THE DARK.
- Add 2 ml cold Washing Buffer (e.g. PBS + 2% Bovine Serum Albumin), gently mix and centrifuge 5 min at 200 X g, 4° C.
- Aspirate and discard supernatant. Gently vortex to break up cell pellet and repeat Step 7.
- Resuspend pellet in 100 µl protein-free, cold PBS and mix well.
- Add 300 µl cold PBS + 2% paraformaldehyde. Store in refrigerator, light protected for up to 10 days. Can skip fixation and use Propidium Iodide at 0.5 µg/ml in Washing Buffer and run within 4 hours.
Indirectly Labeled
- Prepare cells in 200 µg/ml normal IgG of the second antibody species (e.g. goat) at 5 - 10 X 106 cells/ml.
- Start with Ab stock at 300 µg/ml in PBS. Serial dilute six tubes:
10 µl Ab + 20 µl PBS (or 20 µl of previous dilution) = 30 µl
- Aliquot 50 µl of cells in (Falcon #2054) 12 X 75 mm tubes on ice.
- Add 10 µl of each Ab dilution. Also prepare a tube with cells alone (autofluorescence control) and isotype negative control.
- Gently mix incubate 15 - 45 min on ice.
- Add 2 ml cold Washing Buffer (e.g. PBS + 2% BSA), gently mix and centrifuge 5 min at 200 X g, 4° C.
- Aspirate and discard supernatant. Gently vortex to break up cell pellet and repeat Step 6.
- Resuspend cells in 100 µl of appropriately diluted fluorochrome-conjugated second antibody (e.g. goat anti-mouse FITC).
- Incubate 15 - 45 min on ice IN THE DARK.
- Wash twice as above (Steps 6 & 7).
- Resuspend pellet in 100 µl protein-free, cold PBS and mix well.
- Add 300 µl cold PBS + 2% paraformaldehyde. Store in refrigerator, light protected for up to 10 days. Can skip fixation and use Propidium Iodide at 0.5 µg/ml in Washing Buffer and run within 4 hours.
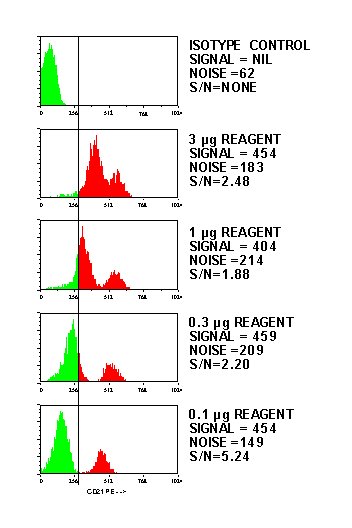
Here's an example of a titration showing the expected decrease in fluorescence with a corresponding increase in the signal to noise ratio:
Reference
Current Protocols in Cytometry, J Wiley & Sons, N.Y., eds. Robinson, J.P. et al, 4.1-4.4, 1997.
YEAST CELL CYCLE
These dyes work better than Propidium Iodide:
SYTOX Green
SYTOX Green is better than PI.
Materials
RNASE
2 mg/ml RNAse A in 50 mM Tris pH 8.0 & 15 mM NaCl
(boil for 5 -10 minutes and allow to cool, aliquot and freeze)
PROTEASE
5 mg/ml pepsin, 4.5 microliter/ml concentrated HCl in H2O
SYTOX GREEN STAINING SOLUTION
1 mM SYTOX Green in 50 mM Tris pH 7.5
Method
- Pellet 107 cells and re-suspend in 1.5 ml H2O.
- Slowly add 3.5 ml 100% EtOH. Fix at least 1 hr RT (or overnight in the refrigerator).
- Wash fixed cells once with 1.0 ml H2O.
- Re-suspend in 0.5 ml RNAse solution and incubate for 2 - 12 hours at 37º C.
- Pellet and re-suspend in protease solution and incubate 20 minutes at 37º C.
- Pellet and re-suspend in 0.5 ml 50 mM Tris pH 7.5.
- Can be stored at 4º C.
- Add 50 - 100 microliters of processed cells to 500 - 900 microliters of SYTOX Green Staining Solution in Falcon® 12 X 75 polystyrene tubes [35]2052, 2054, or 2058.(DO NOT SUBSTITUTE TUBES.)
- Sonicate and analyze.
Reference
Haase SB, Reed SI. Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle. 2002 Mar-Apr;1(2):132-6. PMID: 12429922
SYBR Green I
SYBR Green I is better than PI.
Materials
RNASE
2 mg/ml RNAse A in 50 mM Tris pH 8.0 & 15 mM NaCl
(boil for 5 -10 minutes and allow to cool, aliquot and freeze)
PROTEINASE K
20 mg/ml in H2O
TRIS-EDTA
3.7 g Tris base
121 mg EDTA
in 1 liter H2O pH 8.0
SYBR GREEN I STAINING SOLUTION
1:10 dilution of SYBR Green I in Tris-EDTA with 0.25% (v/v) Triton X-100
SODIUM CITRATE BUFFER
14.7 g sodium citrate in 1 liter H2O pH 7.5
50º C WATERBATH
Method
- Pellet 107 cells and re-suspend in 1.5 ml H2O.
- Slowly add 3.5 ml 100% EtOH. Fix at least 1 hr at Room Temperature (or overnight in the refrigerator).
- Wash fixed cells once with sodium citrate buffer and transfer to Falcon® 12 X 75 polystyrene tubes [35]2052, 2054, or 2058.(DO NOT SUBSTITUTE TUBES.)
- Re-suspend in 0.5 ml RNAse solution and incubate for 1 - 2 hours at 50º C.
- Add 20 microliters Proteinase K solution and incubate 1 hour at 50º C.
- Add 20 - 100 microliters of SYBR Green I Staining Solution and incubate overnight in the refrigerator.
- Sonicate and analyze.
Reference
Fortuna M, Joao Sousa M, Corte-Real M, and Leao C. in Current Protocols in Cytometry, UNIT 11.13.
Other protocols
20X Phosphate Buffered Saline (PBS) Stock Solution
(Dulbucco's w/o Ca+2 & Mg+2)
20X for a longer shelf life than 1X or even 10X.
20X PBS Stock Solution
Dissolve in THIS ORDER (they'll dissolve faster) in 1 liter of H2O:
- 2.88 g Na2HPO4
- 160.00 g NaCl
- 4.00 g KH2PO4
- 4.00 g KCl
Adjust to pH 7.2 with 10 M NaOH. Filter and store at room temperature.
1X PBS Working Solution
- Add 50 ml of 20X PBS Stock Solution to 950 ml glass distilled water.
(Optional) Add 1 g NaN3 and/or 20 g Bovine Serum Album. - Mix well.
- Store at 4°C.
- Use cold buffer for diluting and washing cells.
Erythrocyte Lysis
Erythrocyte Lysing Solution (no preservative) 10X Stock Solution
Dissolve in 1 liter of H2O:
- 89.9 g NH4Cl
- 10.0 g KHCO3
- 370.0 mg tetrasodium EDTA
- Adjust pH to 7.3. Store at 4°C in full, tightly closed 50 ml tubes.
1X Working Solution
Add 5 ml 10X Lysing Stock Solution to 45 ml H2O. Mix well. Store at room temperature for up to one week.
PROCEDURE
- Mix 14 ml Lysing Buffer to 1 ml Blood.
- Incubate for 3 - 5 minutes.
- Centrifuge at 300 x g for 5 minutes at room temperature.
- Aspirate supernatant and resuspend pellet in 5 ml of COLD Phosphate Buffered Saline (PBS).
- Centrifuge at 300 x g for 5 minutes at 2 - 8°C.
- Aspirate supernatant and resuspend pellet in 1 ml of COLD PBS with protein carrier.
Leukocytes via Dextran Sedimentation
- Draw 15 ml blood into a 20 ml syringe containing 0.6 ml 15% Dextran and 0.6 ml 0.25M EDTA (both in 0.9% saline at room temperature).
- Place syringe at 30° angle for 10-30 minutes, then stand upright for another 15-30 minutes at room temperature.
- With a new, bent 18 gauge needle (Monoject #8881-200078), carefully push the leukocyte-rich plasma into a 15 ml centrifuge tube.
- Centrifuge at 300 x g for 5-10 minutes at room temperature.
- Aspirate supernatant and resuspend pelleted cells with 1 ml protein-free buffer (eg. Phosphate Buffered Saline, PBS).
- Lyse erythrocytes for 10 seconds with 9 ml H2O, then add 1 ml of 10X PBS.
OR USE BDIS PROTOCOL (HYPOTONIC NH4CL/KHCO3/EDTA BUFFER) FOR LYSIS INSTEAD:
- To 1 ml cells add 14 ml of 1X Lysing Solution. See Becton Dickinson Source Book Procedures Section 2.11 (or view drawer above: "Erythrocyte Lysis" ) for recipe.
- Mix well and incubate 3-5 minutes at room temperature.
- Centrifuge at 300 x g 5 minutes at room temperature.
- Aspirate supernatant, resuspend cell pellet with 5 ml of COLD buffer.
- Centrifuge at 300 x g 5 minutes at 2° to 8°C.
- Aspirate supernatant, resuspend to 1 ml buffer with protein carrier (eg. Bovine Serum Albumin, BSA).
References
- Tausk F, Fey, M, Gigli, I. J Immumol 143:3295-3302. 1989.
- Moore, JS, Caulkins CE. J Immounol 134:3838-3844, 1985.
- Becton Dickinson Monoclonal Antibodies Source Book, 1995.
Para-Formaldehyde Recipe
For 500 ml 2% Paraformaldehyde Solution
- Allow paraformaldehyde (PFA) powder (Aldrich #30525-89-4 or Sigma P-6148) to come to room temperature (Stored in refrigerator)
- Weigh 10.0 g PFA in fume hood
- Flush container with Argon or Nitrogen to prevent air decomposition of p-formaldehyde.
- Dissolve in 475 ml distilled H2O in 60-70º C water bath on hot plate in fume hood. Do not allow water bath to go over 70ºC (formaldehyde will vaporize!).
- While disolving, label 100 X 4 ml and 7 X 12 ml tubes (or other combinations of useful aliquots) with 2% PFA and the date
- After @ 1 hr add 1 or 2 drops of 5M NaOH. Cloudy suspension will then turn clear.
- Allow to cool to room temperature (@ 2 hours).
- Add 25 ml 20X PBS (see first drawer in "Other Protocols") and adjust pH to 7.3.
- Filter, aliquot into tubes and freeze. Aliquots good for at least 5 years.
Working Diltution
Thawed aliquots are stable at 4º C for up to 2 weeks.
Dilute 1 part 2% PFA to 3 parts cells in PBS (eg 60 µl + 180 µl to yield 0.5% final concentration).
References
- Becton Dickinson Immunocytometry Systems Source Book (1989) 2.10
- Lanier, L.L., and Warner, N.L. (1981) Paraformaldehyde Fixation of Hematopoietic Cells for Quantitative Flow Cytometry (FACS) Analysis. Journal of Immunological Methods 47, 25
Links to other protocol pages
- Cytometry Techniques and Protocols (Purdue University)
- St. Mary's Flow Cytometry Core Facility (UK)
- Molecular Probes Handbook (ThermoFisher Scientific)
- University of Florida Interdisciplinary Center for Biotechnology Research: